ACHAIKI IATRIKI | 2021; 40(4): 217–220
Review
Foteini-Theodora Milidaki, Panagiota Sakellaraki, Efthemia Papakonstantinou, Vasiliki Zolota, Vasiliki Tzelepi
1Department of Pathology, University Hospital of Patras, Patras, Greece
Received: 14 Jul 2021; Accepted: 18 Oct 2021
Corresponding author: Vasiliki Tzelepi, Department of Pathology, University Hospital of Patras, Patras, Greece, Rion Patras, Greece, Tel.: 2613604082, Fax: 2610991810, E-mail: btzelepi@upatras.gr
Key words: Colon cancer, microsatellite instability, KRAS, BRAF, CIMP
![]()
Abstract
Colorectal cancer is a common cause of cancer-related deaths. Significant advances have been made in recent years regarding the understanding of its pathogenesis. Colorectal carcinomas develop through the serial accumulation of genetic and epigenetic events along two pathways: the chromosomal instability pathway, where adenomas are the precursor lesions and APC and KRAS mutations represent early events, and the CpG island methylator pathway, where serrated lesions are the precursor lesions, BRAF or KRAS mutations represent early events and epigenetic MLH1 silencing is a frequent occurrence activating the microsatellite instability pathway (MSI). Carcinomas in patients with familial adenomatous polyposis develop along the chromosomal instability pathway, whereas in Lynch syndrome mutations in mismatch repair genes (that is MLH1, MSH2, MSH6, PMS2) result in microsatellite instability. These developments have important therapeutic implications and testing for the presence of KRAS/BRAF mutations and MSI is recommended in patients with colorectal carcinomas to guide therapeutic decisions in the era of precision medicine.
Introduction
Colorectal cancer (CRC) is the second (women) and third (men) most common malignant neoplasm [1], with its incidence increasing in the last years, especially in low and middle income countries [2], probably due to lifestyle changes. It also represents the third (women) and fourth (men) most common cause of cancer-related death in humans [1]. Increased cancer screening and development of newer therapeutic modalities has resulted in a decrease in mortality rates (in patients older than 50 years) [3]. There has been an increasing understanding of colorectal carcinogenesis pathways in the last two decades; cancer predisposition inherited syndromes, notably familial adenomatous polyposis and Lynch syndrome, that consist 5% of all CRC cases, have played a major role in this progress [4].
Colorectal cancer develops through a stepwise accumulation of genetic and epigenetic abnormalities than enable cells to bypass proliferation control, evade apoptosis, avoid immune destruction, promote angiogenesis, and survive and proliferate at metastatic sites [5]. Along this serial accumulation of genetic and epigenetic events, recognizable lesions are formed (i.e. adenomas, serrated lesions) that progress to more advanced lesions through additional mutations or epigenetic events, a process that requires 10 to 15 years [6]. The molecular pathways that have been linked to the pathogenesis of colorectal carcinoma are the chromosomal instability (CIN) pathway, the microsatellite instability (MSI) pathway, and the CpG island methylator phenotype (CIMP) pathway [7]. A fourth pathway, the polymerase proofreading aberrations pathway is responsible for a minority (<3%) of CRC tumors [3]. In this review we summarize the three major pathways of colorectal carcinoma pathogenesis, with an emphasis on the genes implicated, the associated cancer predisposition syndromes and the therapeutic implications of selected biomarkers. Preneoplastic lesions are also mentioned in brief.
Chromosomal instability (CIN
The CIN pathway consists of gains and losses of whole chromosomes or fractions of them and it usually occurs as a result of mutations in proto-oncogenes or tumour suppressor genes [8,9]. Tumours developed through this pathway are characterized by aneuploidy (abnormal number of chromosomes in a cell, that is 46+/-n) and loss of heterozygosity (LOH, the somatic loss of wild-type alleles, concerning the entire gene and the surrounding chromosomal region). Genes frequently altered in tumours developing through CIN can be divided in three categories, that represent different stages of cell cycle progression: chromosomal segregation, telomere stability and DNA damage response [9–11].
Chromosome segregation results in separation of the duplicated chromosomes into the daughter cells during mitosis by the mitotic spindle [11]. This procedure is briefly paused by the spindle checkpoint, taking place in prometaphase, until all chromosomes have established bipolar connections (bioriented chromosomes) to the mitotic spindle. On instances where specific pairs of sister chromatids are not properly aligned on the metaphase plate, usually due to kinetochores not being properly attached to microtubules or not under enough tension by spindle-pulling forces [12,13] a signal is generated so that anaphase is delayed [14]. This signal is received by a series of spindle-checkpoint proteins, including MAD1, MAD2, BUB3, BUB1, BUBR1, and CENP-E (centrosome protein E). The checkpoint inhibits the APC/C (anaphase-promoting complex/cyclosome) and its coactivator Cdc20, (APC/C(Cdc20)). When all chromosomes are properly attached and aligned, the APC/C complex initiates ubiquitin-dependent degradation of securin and activation of separase, which in turn dissolves the cohesion between sister chromatids, by cleaving a multiprotein complex termed cohesin. Cohesin is responsible for the cohesion between the sister chromatids and plays an important role in chromosome segregation in dividing cells [15].
Mutations in genes that participate in this process lead to errors in segregation and, occasionally, carcinogenesis. More specifically, mutations in kinetochore proteins hRod/KNT, hZw10 and hZwilch/FLJ10036, which contribute to the spindle checkpoint, have been identified in colorectal cancer cases [16]. Aurora kinases, which consist of AURKA (Aurora A), AURKB (Aurora B) and AURKC (Aurora C), are serine/threonine kinases that also participate in regulating the process of chromosomal segregation, each one having their own role and localization [17]. AURKA is associated with centrosome maturation and bipolar spindle formation. When overexpressed, it leads to amplified centrosomes and defective spindle formation. Subsequently, mitosis is inhibited and the incomplete cytokinesis results in multinucleation [9,18,19]. AURKA has been associated with some instances of CIN in colorectal tumours [20]. AURKB is a passenger protein and participates in cytokinesis, chromatid segregation and the modification of histones. Its overexpression relates to advanced stages of colorectal cancer [21].
Telomere dysfunction has also been recognized as a causative factor for CIN. Chromosome ends are protected by telomeres, which are short DNA sequences with their associated proteins. Their main function is to protect the chromosome ends from double-strand breaks, occurring during segregation, and to prevent them from fusing [9,22]. However, after each replication round, a part of telomeric DNA is lost, which leads to telomere shortening (end-replication problem). This occurs as a result of the inability of DNA polymerase to fully form the 3’ end of linear chromosomes. When telomeres reach a critically short length, the cell enters senescence, while the ones that fail to complete this path undergo massive cell death, after entering a crisis-state. Activation of telomerase, the enzyme that is responsible for elongating telomeres, or the ALT (Alternative Lengthening of Telomeres) mechanism, occurs in cells that manage to survive the aforementioned crisis [9].
DNA-damage response (DDR) mechanisms have evolved in order to detect structural DNA alterations, occurring mainly because of environmental agents, reactive oxygen species and spontaneous hydrolysis of nucleotide residues [23,24]. These protein complexes play an important role in identifying an error in a DNA sequence, so that the cell cycle is paused, allowing the damage to be repaired, or, in cases where the damage is beyond repair, halting cell’s growth or initiating its apoptosis.
As expected, cells with abnormal DDR are more sensitive to DNA damaging sources, and exhibit genomic instability. Specifically, some of the genes whose protein products participate in these signalling and repair processes have been reported to relate to certain syndromes that predispose humans to cancers. Some examples are ATM (ataxia telangiectasia mutated) and ATR (ataxia telangiectasia and Rad3-related) protein kinases, involved in Louis-Bar syndrome and Seckel syndrome respectively, BRCA1, BRCA2 proteins, whose mutations lead to hereditary ovarian and breast cancer and WRN (Werner syndrome protein) linked to Werner syndrome. Last but not least, mutations in TP53 protein have been causatively related to Li-Fraumeni syndrome, which has been linked to CIN in colorectal cancer [9,25].
The Wnt/β-catenin pathway plays an important role in the pathogenesis of colorectal cancer, primarily affected by mutations of the APC (Adenomatous Polyposis Coli) gene (5q21). The protein encoded by this gene participates in many cell functions, but especially noteworthy is its participation in the Wnt signalling pathway, as it is crucial for tumorigenesis. The purpose of the Wnt pathway is to control the translocation of β-catenin to the cell nucleus. After its migration, β-catenin, interacts with the TCF/LEF (T-cell factor/lymphoid enhancer factor) family of transcription factors and, thus, transcriptionally activates various genes, which are involved in cancer growth. When Wnt ligands are absent, a complex consisting of APC, Axin, CK1 (casein kinase) and GSK-3β (glycogen synthase kinase) mediates β-catenin degradation. More specifically, the amino terminal area of β-catenin is phosphorylated by the two kinases of the complex, which allows β-Trcp, a protein with ubiquitin ligase activity, to recognize it. Subsequently, β-catenin is ubiquitinated and subjected to degradation by proteasomes [26].
When a Wnt ligand is present, it binds to FZD (Frizzled) and its co-receptors LRP5 or LRP6 (low-density lipoprotein). Subsequently, a complex called ‘signalosome’ is formed, which interacts with the Dvl protein (Dishevelled) and causes LRP6 phosphorylation and Axin complex activation. This mechanism inhibits β-catenin phosphorylation. β-catenin is, then, translocated to the nucleus where it participates in transcriptional gene activation [26]. When APC is mutated, the β-catenin degradation complex is not formed and, the pathway is constantly activated, even in the absence of a signal, leading to uncontrolled cell proliferation [26–28].
It has been reported that APC mutations are an early event in colon carcinogenesis. APC appears somatically mutated in 5% of dysplastic aberrant crypt foci, 30%–70% of sporadic adenomas and 70% of sporadic tumors [9,29–31]. Another, albeit not as common, mechanism responsible for APC gene inactivation is the hypermethylation of the gene’s promoter (seen in 18% of primary colorectal carcinomas and adenomas) [32]. In addition, germline mutations of APC have been directly linked to FAP (Familial Adenomatous Polyposis), an inherited colon cancer predisposition syndrome (see below) [33,34].
Apart from APC, gain-of-function mutations have been reported in the gene encoding β-catenin (CTNNB1) in 50% of colorectal tumours without APC mutations [32,35]. It has been shown that CTNNB1 mutations are more frequent in small adenomas (12.5%) than they are in large ones (2.4%) and invasive cancers (1.4%), [36], a finding which suggests that they are not as prone to induce malignancy as APC mutations. Finally, AXIN and AXIN2/conductin gene mutations have been found in CRC, albeit not in those developing through the CIN pathway, but rather those with an MSI high phenotype (see below) [37,38].
In the adenoma to carcinoma sequence, in the CIN pathway of CRC carcinogenesis, a relatively early event following APC mutations and WNT pathway activation, with significant clinical implications, is KRAS mutation [9]. Proteins that belong to the RAS family (three isoforms; KRAS, NRAS, and HRAS with >80% homology) possess the ability to bind guanosine triphosphate (GTP) and guanosine diphosphate (GDP), cycling between their active and inactive alternative states, respectively [39]. Their role is to mediate a number of signalling pathways, so that extracellular signals are transduced to the cell nucleus, and enhance gene transcription, leading to initiation/regulation of cellular procedures, like cell proliferation and growth, differentiation and migration [40]
RAS, in its activated form, is involved in many signal transduction pathways, including the RAS/RAF/MAPK pathway (also known as the mitogen-activated protein kinase (MAPK) cascade) and the PI3K/AKT pathway [40]. These pathways participate in regulating cell cycle, migration and apoptosis, tissue healing and angiogenesis, important hallmarks of carcinogenesis [5].
Following ligand (i.e. epidermal growth factor-EGF) binding to its receptor (i.e. epidermal growth factor receptor-EGFR), the receptor is dimerized, auto-phosphorylated and activated, thereby activating adaptor proteins which enable RAS to exchange its GDP with GTP. GTP-bound RAS interacts with RAF and activates a phosphorylation cascade. RAF as a family consists of three serine/threonine kinases, A-RAF, B-RAF, and C-RAF (first recognized as retroviral oncogenes in the avian retrovirus Mill Hill 2 (MH2), and the murine sarcoma virus (MSV) 3611 isolate) [41] which are activated by RAS-GTP and in turn, phosphorylate and activate MEK1 and MEK2. The latter are MAPKs (Mitogen-Activated Protein Kinases), four different kinds of which have been recognized: ERK, c-Jun N-terminal kinase (JNK), ERK5 and p38 MAPK (p38) [42]. MEK1 and MEK2, which are characterized by substrate specificity, catalyse the phosphorylation of ERK1 and ERK2, which proceed to phosphorylate multiple substrates (because as opposed to their activators, they have a wider specificity), leading to the regulation of several transcription factors and, as a result the expression of multiple genes [9,43,44].
KRAS is mutated in 30-50% of colorectal cancers [9,45] and is generally considered to be one of the oncogenes that are most frequently mutated in human cancers (also frequently mutated in carcinomas of the lung, pancreas, breast, and oesophagus) [46–49]. In colon cancer, KRAS mutations are a relatively common event as they have been identified in 60-95% of aberrant crypt foci (the earliest morphologic manifestation of adenomas) [9,50,51]. KRAS mutations are not limited to carcinomas developing through the chromosomal instability pathway, but are an early event in serrated carcinogenesis too (through a traditional serrated adenoma precancerous lesion) (see below).
KRAS mutations usually occur in exon 2, followed by mutations in exons 3 and 4. Most of its mutations consist of amino acid substitutions (caused by single nucleotide point mutations) in codons 12 and 13 of exon 2, amounting for 88% of recurrent mutations in all types of cancers. Mutations may also appear in codons 59 and 61 of exon 3, and in codons 117 and 146 of exon 4, albeit less frequently. On the other hand, NRAS and HRAS mutations are much less frequent than KRAS mutations and are usually located in codons 61 and 59 (exon 3), followed by codons 12 and 13 of exon 2 and 117 and 146 of exon 146 [43,52]. These alterations have the same effect, diminishing the molecule’s endogenous GTPase activity and inducing a constant GTP-bound state, leading to continuous activation of the cascade and finally promoting cell survival, proliferation and migration and inducing carcinogenesis.
A therapeutic choice for metastatic colorectal carcinoma is EGFR inhibition. In contrast to lung cancer, EGFR mutations are not common in colorectal cancer and their presence does not predict therapeutic response. In contrast, the presence of KRAS/NRAS mutations has a negative predictive role as they predict lack of response to EGFR targeting therapy [53], since activation of the pathway is due to a genetic event downstream of the receptor. Initially, exon 2 KRAS mutations were identified as predictive [52,54,55], but further research [56,57] has shown that all the mutations mentioned above are associated with lack of response to anti-EGFR targeting therapy (monoclonal antibodies cetuximab and panitumumab, targeting the extracellular domain of the receptor). Thus, current guidelines from the American Society of Clinical Oncologists, the College of American Pathologists, and the Association for Molecular Pathology recommend that extended ras analysis, including KRAS [exons 2 (Codons 12,13), 3 (codons 59, 61) and 4 (codons 117, 146)] and NRAS [exons 2 (Codons 12,13), 3 (codons 59, 61) and 4 (codons 117, 146)], should be performed to all patients with metastatic colorectal carcinoma considered for anti-EGFR therapy [58,59]. Only patients with wild-type KRAS are candidates for this type of therapies [52,60] as those have a higher probability of responding to the treatment. This way patients not likely to respond are excluded and saved unnecessary toxicity, and cost. An estimate of 7500$ per patient is saved when these predictive markers are used in therapy selection [61,62].
TP53 is another gene frequently mutated in tumours associated with CIN, albeit this happens relatively late in the pathway. This gene encodes a nuclear transcription factor which acts as a tumour suppressor, inducing cell cycle arrest when the DNA appears damaged. That way, DNA can be repaired or, in case of irreversible damage, the cell is led to apoptosis. Loss of function mutations in TP53 have been reported in more than 50% of cancers, so TP53 dysfunction is generally considered a hallmark in human tumours [63]. Regarding colorectal cancer, loss of function in TP53 has been increasingly found with progression of the lesion as it is seen in 4%–26% of adenomas, 50% of early carcinomas developing in adenomas, and in 50%–75% of late carcinomas [9,64], making TP53 mutations a defining event in the adenoma to carcinoma progression. Most of its mutations are missense: transitions of GC to AT principally occurring in five hotspot codons (175, 245, 248, 273, and 282) [65].
Other abnormalities frequently encountered in CRC associated with CIN are COX-2 overexpression leading to overexpression of its product, PGE2, which regulates proliferation, tumorigenesis and angiogenesis [9,66], and loss of 18q (where SMAD2 and SMAD4, mediators of the TGFb pathway are located) [67,68]. Mutations in PIK3CA leading to its activation, occur late in the adenoma to carcinoma progression in a small proportion of cancers [69] and even though there are some reports for a positive predictive function of their presence in regards to aspirin effect in reducing CRC recurrence [70,71], data are conflicting [72] and current guidelines do not include PIK3CA mutational analysis as necessary for CRC patients [58].
Familial Adenomatous Polyposis
Familial adenomatous polyposis (FAP) is an autosomal dominant syndrome, caused by germline APC (5q21) mutations (frequency 1 in 6,850 to 29,000 people) [73]. Patients with FAP develop hundreds to thousands of adenomas, morphologically indistinguishable from sporadic adenomas. One or more adenomas will eventually progress to carcinoma and the lifetime probability of developing colorectal cancer in FAP individuals is 100%, unless a colectomy is performed (usually at an age between 15 and 25 years old) [74]. This syndrome presents with various degrees of penetrance and, thus, different phenotypes, which is not surprising considering the fact that causative mutations can occur at different loci in the gene and that environmental factors may alter the disease phenotype [74,75].
The hallmark feature of this disease is the development of adenomatous polyps along the GI tract beginning in early adolescence, with a rapid increase in number and size with age, and progression to colorectal cancer by the fourth decade [33]. The adenomas that develop in FAP are histologically similar to sporadic adenomas (Figure 1) [76]. Almost 75% of patients suffering from FAP have already developed colorectal carcinoma by the age of 30 [73].
Lesions can develop not only in the colon and rectum, but also throughout the GI tract. In the stomach mostly fundic gland polyps are seen, usually benign and morphologically similar to their non-syndromic counterparts, but, unlike sporadic polyps, exhibiting dysplasia in 25% of the cases [77]. In the small intestine adenomas are seen in 30-70% of patients with FAP, most commonly in the periampullary region of the duodenum [74,78]. Adenomas vary in size, from 1mm to > 1cm, and their number ranges from 100 to more than 5.000. Attenuated FAP is characterized by <100 polyps, increased risk of CRC development (albeit a little less than classic FAP and at an older age) and mutations involving the 5’ or 3’ part of the gene [75]
Patients with FAP occasionally present extracolonic manifestations [74,75] in the bones (osteomas in 65-80% of patients), teeth (dental abnormalities found in 30-75% of patients: impacted or unerupted teeth, tooth ankylosis, congenitally missing teeth, supernumerary teeth, compound odontomas), retina (Congenital Hypertrophy of Retinal Pigment Epithelium, CHRPE, the most common and earliest extraintestinal manifestation of FAP), thyroid (cribriform-morular variant of papillary thyroid carcinoma), liver (hepatoblastoma), central nervous system (brain tumours in general, especially patients with APC mutations between codons 697 and 1224) [79].
After FAP has been confirmed (clinical criteria or APC mutation confirmed by genetic testing) [33], treatment can be surgical (colectomy with or without proctectomy) or non-surgical (NSAIDS, COX-2 inhibitors). However, delaying the former with the use of medication that reduces the number of adenomas has limited results [75]. Annual screening is recommended even after the patient has undergone surgery [73,74].
MSI (Microsatellite instability)
Microsatellites represent repetitive DNA sequences (1 to 8 nucleotide units long), found throughout the human genome [80,81]. Alterations in their length are caused by DNA polymerase slippage, resulting in insertion or deletion of base pairs, thus, altering the number of repeats [81–83]. Responsible for recognition and correction of this type of errors (insertion/deletion mispairs) is the DNA mismatch repair (MMR) system, which also repairs base mismatches [84].
The MMR system comprises of several proteins: hMutSα (formed by heterodimerization of MSH2 with MSH6) and hMutSβ (MSH2-MSH3 heterodimer) recognize mismatches and single base insertions/deletions loops or 2-8 bases insertions/deletions loops, respectively. Then, they recruit hMutLα (MLH1-PMS2 heterodimer), hMutLß (MLH1-PMS1 heterodimer) or hMutLγ (MLH1-MLH3 heterodimer), along with replication factors and other proteins [83–85]. EXO1, single-strand DNA-binding protein RPA, proliferating cellular nuclear antigen (PCNA), DNA polymerase d (pol d), and DNA ligase I, are also involved in this process and mediate the excision of the most recently synthesized helix (the one containing the error), its re-synthesis (with the correct sequence) and ligation of the new helix with the rest of the DNA [84].
Loss of action of components of the DNA MMR system (known as MMR deficiency), is associated with a propensity for multiple point mutations across the genome (hypermutability), as well as insertions and deletions in microsatellite sequences, the latter accounting for the name of this state as microsatellite instability (MSI or MSI-high) [86,87]. Almost 15% of all colorectal cancers have been reported to display MMR deficiency and MSI [80,86]. Defects in this system occur as a result of germline mutations in MMR genes (in Lynch syndrome), or epigenetic inactivation by promoter hypermethylation (in sporadic MSI high tumours).
Screening patients for MSI in colorectal carcinomas and determining MMR functionality is important, as it can help identify individuals with Lynch syndrome. This has profound consequences not only for the patients themselves (increased colon surveillance for the development of subsequent tumours, screening for the development of tumours in other organs frequently affected in this syndrome), but also for their family members that may also be affected and need to be enrolled in screening programs [82,88]. In addition, MSI high carcinomas are associated with better prognosis [89] and according to the 2021 NCCN guidelines adjuvant treatment is not needed for patients with MSI high stage II tumours [90]. However, if adjuvant therapy is needed, MSI-H tumours do not respond well to therapy based on traditional cytotoxic agents (such as 5-FU, oxaliplatin, irinotecan) and different regimens should be used. Lastly, in the metastatic stage, MSI can predict response to immune checkpoint inhibitors [80,81] such as anti-PD-1 antibodies (nivolumab, pembrolizumab) and the anti-CTLA-4 antibody (ipilimumab). Consequently, pembrolizumab has been FDA approved for use as first line treatment in patients with dMMR/MSI-H CRC [91,92]. Nivolumab (alone or in combination with ipilimumab) [93] has been FDA approved for use in patients with dMMR/MSI-H CRCs that have progressed following treatment.
Tumours that are MSI-H have been linked to certain characteristics, such as location in the proximal part of the colon, mucinous (i.e mucins pools with neoplastic cells floating within them) or medullary (solid nested or trabecular syncytial growth with tumor infiltrating lymphocytes) histology, poor differentiation, lower rates of KRAS and TP53 mutations, and increased immune cell infiltrates (sometimes with a Crohn-like reaction) [94,95]. However, the predictive value of the histologic characteristics in regards to MSI status is rather low and, thus, histology is no longer used to guide decisions regarding MSI testing [96]. All newly diagnosed colorectal cancers, regardless of family history, should be subjected to MMR or MSI testing, according to the College of American Pathologists, the American Society for Clinical Pathology, the Association of Molecular Pathology, and the American Society of Clinical Oncology [58] and the National Comprehensive Cancer Network (NCCN) [90].
MSI and MMR status can be detected by various methods: MSI-PCR, immunohistochemistry and next-generation sequencing (NGS). Regarding MSI-PCR, the National Cancer Institute (NCI) has recommended five microsatellite sequences as markers, known as the Bethesda panel: BAT25, BAT26, D2S123, D5S346 and D17S250 [97], albeit additional microsatellite markers are now commercially available. In principle, the length of each marker is compared between tumour tissue and normal tissue. Tumours can therefore be classified in three categories, based on MSI status: MSI-high (MSI-H), indicating a difference in the length of two or more of the five markers in tumour DNA, MSI-low (MSI-L), when only one marker exhibits a difference in its length, and microsatellite stable (MSS), when all markers have the same length in tumour and healthy tissue [81,98,99].
Another method for determining MSI status is detecting the absence of the expression of one or more of the four MMR proteins (MLH1, MSH2, MSH6 and PMS2) with immunohistochemistry (IHC), a state known as defective MMR (dMMR). Because these proteins function as heterodimers, PMS2 and MSH6 are usually unstable without MLH1 and MSH2 expression (their dimer partners), respectively. Thus, when MLH1 expression is lost, PMS2 (its dimer partner) is also lost (same with MSH2 and MSH6) (Figure 2). In contrast, MLH1 and MSH2 are stable even when PMS2 and MSH6 are absent. Therefore, loss of PMS2 and MSH6 is not accompanied by loss of their partners MLH1 and MSH2, respectively [99,100].
Comparison between the two methods (MSI-PCR and immunohistochemistry) has shown a high level of concordance [99,101]. IHC advantages are that it is fast, low-cost and readily available in most laboratories, it has low requirements in terms of tissue quantity and is the preferred method in cases with low tumour content (i.e. intense inflammation) [102]. In addition, it can specifically indicate which MMR gene is mutated. However, in up to 10% of the cases, mutations in the MMR genes although affecting their function (thus, the cells are dMMR), they do not affect the protein’s expression (thus, immunohistochemistry is falsely positive) [103]. In addition, technical issues and previous therapy may affect the IHC results [102]. Nonetheless, both methods are crucial as they complement each other in regards to recognizing defective MMR [104]. Newer techniques, such as next generation sequencing are also effective in determining MSI status, with comparable results to PCR and immunohistochemistry [105] and the advantages of simultaneous analysis of multiple genetic aberrations [106,107], and, in some instances, not requiring paired normal tissue [108]. NGS challenges include high cost, increased technical demands, difficulties in data interpretation and poor diagnostic yield in samples with poor DNA quality, but technology is continually improving [109,110] and, in the future, it may lead to its more widespread use.
Lynch syndrome
Lynch syndrome, formerly known as hereditary nonpolyposis colorectal cancer (HNPCC, a term not currently preferred), is inherited by an autosomal dominant pattern and is characterized by a high risk of developing various types of tumours, especially, colorectal and endometrial carcinomas [111,112]. Other types of tumours that have a high probability of developing in individuals with Lynch syndrome are carcinomas of the breast, ovary, stomach, pancreas, small bowel, liver, bile duct, kidney, prostate, and urinary tract. In addition, brain tumours, namely medulloblastomas, and certain types of skin cancers develop in variants of the disease (Turcot and Muir-Torre syndrome respectively [88], the former also termed constitutional mismatch repair deficiency syndrome and is usually seen with homozygous mutations of one of the MMR genes [113].
Inherited alterations in MSH2 (40%) and MLH1 (30%) are responsible for the largest proportion of Lynch syndrome cases, followed by PMS2, and MSH6. Another genetic event recognized as causative of Lynch syndrome is the deletion of the EPCAM gene, resulting in MSH2 methylation and, thus, loss of its expression [114]. There are some differences in the phenotype of the disease depending on the affected gene, in regards of the risk and type of cancer that is developed [76]. The risk of cancer development is higher in MSH2 and MLH1 mutations, followed by MSH6 mutations and the least when PMS2 is affected. In addition, the risk of extracolonic manifestations is null with certain EPCAM deletions [76].
CIMP (CpG island methylator phenotype)
CpG islands are regions where CpG dinucleotide (a cytosine nucleotide followed by a guanine nucleotide) clustering is observed and are commonly found in gene promoters. DNA methyltransferases mediate the transfer of a methyl group to the C-5 position of the cytosine ring of DNA, resulting in CpG methylation which negatively regulates gene expression [115]. The opposite process, DNA demethylation, results in increased gene transcription. DNA methylation is an epigenetic mechanism of gene expression regulation. Epigenetic refers to the change in gene expression without any alteration in DNA sequence [116]. Tumours developing through this pathway are believed to harbour a methylator phenotype, meaning that there is a progressive increase in the methylation of CpG islands, leading to tumour suppressor gene silencing and, thus, tumorigenesis. An overlap with the MSI pathway is observed, as one of the genes frequently (although not always) undergoing hypermethylation in CIMP high carcinomas is MLH1 [117–119]. In fact, in sporadic colorectal cancer, the MSI phenotype, arises as a result of epigenetic silencing of the MLH1 gene by aberrant methylation of CpG islands in its promoter region. Reportedly, epigenetic silencing of MLH1 has been documented in more than 95% of MSI-H (MSI high) sporadic carcinomas [81,82,84,111,120].
Approximately 20–30% of all CRCs exhibit the CIMP phenotype and they are classified in three categories based on their hymermethylation level: low (CIMP-L), high (CIMP-H) or negative (CIMP-0) [121]. Two panels of genes are now widely used to investigate the CIMP status of tumours (p16, hMLH1, MINT1, MINT2 and MINT31, described by Toyota and CACNA1G, IGF2, NEUROG1, RUNX3, and SOCS1 described by Weisenberg) [121,122]. CIMP-H is characterized by activation of the WNΤ/β-catenin pathway, probably induced by non-APC mutations [121], frequent BRAF mutations and MLH1 methylation, with TP53 mutations rarely encountered. On the other hand, CIMP-0 exhibits a high TP53 mutation rate, while CIMP-L is usually associated with KRAS mutations.
Serrated polyps, characterized by a saw-toothed appearance under the microscope (epithelial infolding) are the precancerous lesions in tumours developing through this pathway, hence, also known as the serrated pathway. They are further classified in three categories based on their morphology: hyperplastic polyps, sessile serrated lesions and traditional serrated adenomas (TSAs) or polyps. Most of colorectal cancers with serrated lesions as precursors have been reported to harbour BRAF mutations (with KRAS being less frequently mutated) and have been connected with sporadic MSI and CpG island methylator phenotype (CIMP) [117,121].
Based on their morphology, polypoid lesions have also been associated with specific mutations and CIMP category. Hyperplastic polyps (further classified into microvesicular HP, goblet cell HP and mucin poor HP) usually harbour a V600E mutation in BRAF and belong to the CIMP-H category. BRAF is often mutated in sessile serrated lesions which have been characterized as MSS and CIMP-H with an unmethylated MLH1. The development of dysplasia coincides with the appearance of MSI-H phenotype, MLH1 methylation and higher risk of progression (as mentioned above). As for traditional serrated adenomas or polyps, they often present with KRAS or BRAF mutations, and can either be CIMP-L or CIMP-H, and are MSS [121].
More specifically, the pathway usually starts with BRAF mutations (V600E point mutation) [123], which lead to continuous signalling in the RAS/RAF/MAPK pathway (described in previous paragraphs). BRAF belongs to the RAF (rapidly accelerated fibrosarcoma) family of kinases, originally identified through the cloning of a viral mouse gene that induced transformation of NIH3T3 cells. BRAF is a non-receptor serine-threonine kinase that is located downstream of KRAS. Once activated, (from KRAS) it participates in phosphorylation cascades of the MAPK pathway and, thus, transcriptional activation of genes involved in cell growth, proliferation, survival and migration. When mutated, it is locked in its active form and mediates continuous activation of the pathway [124]. However, after the first wave of proliferation, that results in the development of hyperplastic polyps, one of the earliest manifestations of this pathway, the cell reaches a senescence state, and may remain there for a very long period; in fact, it may never progress. Cell cycle regulators, such as p53 and p16INK4a, play an important role in this oncogene-induced senescence [125], as they halt further proliferation. In some lesions however, silencing of these molecules or their regulators, for example IGFBP7, may ensue, resulting in cell’s escape from senescence and in following bursts of proliferation [117]. A sessile serrated lesion is the morphologic analogous of these molecular events. Again, the lesion may remain stable for a long period, until MLH1 is hypermethylated and dysplasia is encountered morphologically (sessile serrated lesion with dysplasia) (Figure 1). From this stage, evolution of the lesion to invasive cancer (through additional epigenetic events) is usually quicker than in the previous stages and BRAF mutated, CIMP-H, MSI-H (sporadic MSI-H) carcinomas develop (Figure 2 and 3) [121]. In some cases, the pathway may progress though epigenetic silencing of alternative genes (not MLH1), and then, BRAF mutated, CIMP-H, MSS carcinomas develop [121,126,127].

Figure 1. Preneoplastic lesions of the colon. Tubular adenomas develop through the CIN pathway. Sessile serrated lesion is characterized by crypt serration that extends to their base. MLH1 promoter hypermethylation and protein expression loss is characterized by dysplasia morphologically. Traditional serrated polyp with the characteristic villous architecture and ectopic crypt formation. This lesion harboured a KRAS exon 2 mutation (G12X).
BRAF mutations are seen in 5-15% of the patients with CRC, with V600E being the most common and characterized by the substitution of valine by gutamic acid at the 600 position (located at the activation site of the molecule). BRAF mutations are more common in female patients, in tumours located in the right side of the colon and in MSI-H carcinomas [128]. Testing for BRAF mutations is necessary for all MSI-H carcinomas (their absence indicates that the tumour has developed in the setting of Lynch syndrome and should prompt germline genetic testing) [33]. BRAF mutations are associated with worse prognosis (in MSS tumours) and low response to EGFR targeting therapy [128,129]. Based on the results of the BEACON trial [130], double inhibition of BRAF (encorafenib) and EGFR (cetuximab/panitumumab) is a therapeutic option for BRAF V600E mutant CRC after prior treatment [90].
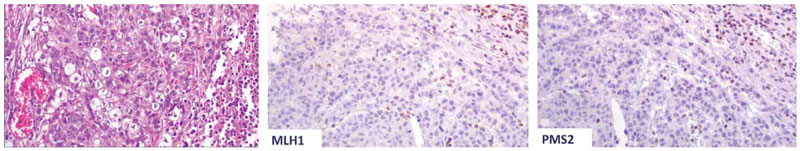
Figure 2. This tumour from a 75-year-old female patient was located in the cecum, and displayed poor differentiation and advanced T stage (T4b) histologically. Loss of both MLH1 and PMS2 was seen immunohistochemically. A BRAF V600E mutation was detected. This is a prototype case of sporadic MSI-H CRC (original magnification X20).
In some instances, where the initiating event may be a KRAS (instead of BRAF) mutation, precancerous lesions are characterized as traditional serrated adenomas (Figure 1), methylguanine methyltransferase (MGMT) gene is methylated [131] and carcinomas that develop have a KRAS mutated, CIMP-L, MSS/MSI-L molecular phenotype [121,127].
CRCs have also been classified into five molecular subtypes [132,133], according to their MSI-CIMP status and the mutational profiles of KRAS and BRAF: (1) type 1: MSI+, CIMP+, BRAF– mutated, KRAS- wildtype; (2) type 2: MSI−, CIMP+, BRAF– mutated, KRAS- wildtype; (3) type 3: MSI−, CIMP−, BRAF– wildtype, KRAS- mutated; (4) type 4: MSI−, CIMP−, BRAF– wildtype, KRAS- wildtype; and (5) type 5: MSI+, CIMP−, BRAF– wildtype, KRAS– wildtype. These distinct categories also exhibit a different prognosis, with type 1 exhibiting the best, type 2 the worst and 5-4-3, with this order, having an intermediate prognosis [126]

Figure 3. The serrated pathway of carcinogenesis. Poorly differentiated carcinoma developing in a sessile serrated lesion with dysplasia. Note the loss of MLH1 in the dysplastic and neoplastic epithelium while residual sessile serrated lesion retains its expression.
Conclusion
This review aims to describe the molecular pathways that have been implicated in the pathogenesis of colorectal carcinoma. As this type of cancer still comprises a major health risk nowadays, it is important to understand and analyse the underlying mechanisms, not only because they offer insights regarding the pathogenesis of the disease, but also because some of them have predictive and prognostic implications and form the basis for personalized therapy in CRC patients.
Conflict of interest disclosure
None to declare
Declaration of funding sources
None to declare
Author contributions
Foteini-Theodora Milidaki: conception and design; analysis and interpretation of the data; drafting of the article; final approval of the article; Panagiota Sakellaraki: analysis and interpretation of the data; final approval of the article; Efthemia Papakonstantinou: critical revision of the article for important intellectual content; final approval of the article; Vasiliki Zolota: analysis and interpretation of the data; critical revision of the article for important intellectual content; final approval of the article; Vasiliki Tzelepi: conception and design; analysis and interpretation of the data; drafting of the article; critical revision of the article for important intellectual content; final approval of the article.
References
1. Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J cancer. 2019;144(8):1941–53.
2. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66(4):683–91.
3. Nagtegaal ID, Arends MJ, Odze RD. Tumors of the colon and rectum. In: Arends MJ, Fukayama M, Klimstra DS, Lam AKY, Nagtegaal ID, Odze RD, et al., editors. Digestive system tumors. 5th ed. Lyon: IARC Publications; 2019 Digestive system tumors. 5th ed.
4. Recio-Boiles A, Cagir B. Colon Cancer [Internet]. StatPearls. 2021 StatPearls.
5. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144(5):646–74.
6. Chen C-D, Yen M-F, Wang W-M, Wong J-M, Chen T-H. A case–cohort study for the disease natural history of adenoma–carcinoma and de novo carcinoma and surveillance of colon and rectum after polypectomy: implication for efficacy of colonoscopy. Br J Cancer. 2003;88(12):1866–73.
7. Requena DO, Garcia-Buitrago M. Molecular Insights Into Colorectal Carcinoma. Arch Med Res. 2020;51(8):839–44.
8. Geigl JB, Obenauf AC, Schwarzbraun T, Speicher MR. Defining “chromosomal instability.” Trends Genet. 2008;24(2):64–9.
9. Pino MS, Chung DC. The Chromosomal Instability Pathway in Colon Cancer. Gastroenterology. 2010;138(6):2059–72.
10. Ryland GL, Doyle MA, Goode D, Boyle SE, Choong DYH, Rowley SM, et al. Loss of heterozygosity: What is it good for? BMC Med Genomics. 2015;8(1):1–12.
11. Santaguida S, Amon A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat Rev Mol Cell Biol. 2015;16(8):473–85.
12. Rieder CL, Cole RW, Khodjakov A, Sluder G. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol. 1995;130(4):941–8.
13. Stern BM, Murray AW. Lack of tension at kinetochores activates the spindle checkpoint in budding yeast. Curr Biol. 2001;11(18):1462–7.
14. Herzog F, Primorac I, Dube P, Lenart P, Sander B, Mechtler K, et al. Structure of the Anaphase-Promoting Complex/Cyclosome Interacting with a Mitotic Checkpoint Complex. Science. 2009;323(5920):1477–81.
15. Peters JM, Tedeschi A, Schmitz J. The cohesin complex and its roles in chromosome biology. Genes Dev. 2008;22(22):3089–114.
16. Wang Z, Cummins JM, Shen D, Cahill DP, Jallepalli P V., Wang TL, et al. Three Classes of Genes Mutated in Colorectal Cancers with Chromosomal Instability. Cancer Res. 2004;64(9):2998–3001.
17. Tang A, Gao K, Chu L, Zhang R, Yang J, Zheng J. Aurora kinases: Novel therapy targets in cancers. Oncotarget. 2017;8(14):23937–54.
18. Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell. 2003;3(1):51–62.
19. Crane R, Gadea B, Littlepage L, Wu H, Ruderman J V. Aurora A, meiosis and mitosis. Biol Cell. 2004;96(3):215–29.
20. Ewart-Toland A, Briassouli P, De Koning JP, Mao JH, Yuan J, Chan F, et al. Identification of Stk6/STK15 as a candidate low-penetrance tumor-susceptibility gene in mouse and human. Nat Genet. 2003;34(4):403–12.
21. Katayama H, Ota T, Jisaki F, Ueda Y, Tanaka T, Odashima S. Mitotic Kinase Expression and Colorectal Cancer Progression Masaaki Tatsuka Loss of chromosomal integrity as well as genomic stability is considered. J Natl Cancer Inst. 1999;91(13):1–2.
22. Murnane JP. Telomere dysfunction and chromosome instability. Mutat Res – Fundam Mol Mech Mutagen. 2012;730(1–2):28–36.
23. Friedberg EC, Aguilera A, Gellert M, Hanawalt PC, Hays JB, Lehmann AR, et al. DNA repair: From molecular mechanism to human disease. DNA Repair (Amst). 2006;5(8):986–96.
24. Lindahl T, Prigent C, Barnes DE, Lehmann AR, Satoh MS, Roberts E, et al. DNA Joining in Mammalian Cells. Cold Spring Harb Symp Quant Biol. 1993;58:619–24.
25. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–8.
26. MacDonald BT, Tamai K, He X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev Cell. 2009;17(1):9–26.
27. He X, Semenov M, Tamai K, Zeng X. LDL receptor-related proteins 5 and 6 in Wnt/β-catenin signaling: Arrows point the way. Development. 2004;131(8):1663–77.
28. Parker TW, Neufeld KL. APC controls Wnt-induced β-catenin destruction complex recruitment in human colonocytes. Sci Rep. 2020;10(1):1–14.
29. Cottrell S, Bodmer WF, Bicknell D, Kaklamanis L. Molecular analysis of APC mutations in familial adenomatous polyposis and sporadic colon carcinomas. Lancet. 1992;340(8820):626–30.
30. Otori K, Konishi M, Sugiyama K, Hasebe T, Shimoda T, Kikuchi-Yanoshita R, et al. Infrequent somatic mutation of the adenomatous polyposis coli gene in aberrant crypt foci of human colon tissue. Cancer. 1998;83(5):896–900.
31. Miyaki M, Konishi M, Kikuchi-Yanoshita R, Enomoto M, Igari T, Tanaka K, et al. Characteristics of Somatic Mutation of the Adenomatous Polyposis Coli Gene in Colorectal Tumors. Cancer Res. 1994;54(11):3011–20.
32. Esteller M, Sparks A, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, et al. Analysis of adenomatous polyposis coli promoter hypermethylation in human cancer. Cancer Res. 2000;60(16):4366–71.
33. Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110(2):223–62.
34. Campos FG. Surgical treatment of familial adenomatous polyposis: Dilemmas and current recommendations. World J Gastroenterol. 2014;.20(44):16620.
35. Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/β-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998;58(6):1130–4.
36. Samowitz WS, Powers MD, Spirio LN, Nollet F, Van Roy F, Slattery ML. Β-Catenin Mutations Are More Frequent in Small Colorectal Adenomas Than in Larger Adenomas and Invasive Carcinomas. Cancer Res. 1999;59(7):1442–4.
37. Krings M, Capelli C, Tschentscher F, Geisert H, Meyer S, Haeseler A Von, et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair. Nat Genet. 2000;26(2):146–7.
38. Shimizu Y, Ikeda S, Fujimori M, Kodama S, Nakahara M, Okajima M, et al. Frequent alterations in the Wnt signaling pathway in colorectal cancer with microsatellite instability. Genes Chromosom Cancer. 2002;33(1):73–81.
39. Roskoski R. RAF protein-serine/threonine kinases: Structure and regulation. Biochem Biophys Res Commun. 2010;399(3):313–7.
40. Roskoski R. RAF protein-serine/threonine kinases: Structure and regulation. Biochem Biophys Res Commun. 2010;399(3):313–7.
41. Zebisch A, Troppmair J. Back to the roots: The remarkable RAF oncogene story. Cell Mol Life Sci. 2006;63(11):1314–30.
42. Li L, Zhao GD, Shi Z, Qi LL, Zhou LY, Fu ZX. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC (Review). Oncol Lett. 2016;12(5):3045–50.
43. Molina JR, Adjei AA. The Ras/Raf/MAPK Pathway. J Thorac Oncol. 2006;1(1):7–9.
44. Simanshu DK, Nissley D V., McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell. 2017;170(1):17–33.
45. Santini D, Loupakis F, Vincenzi B, Floriani I, Stasi I, Canestrari E, et al. High Concordance of KRAS Status Between Primary Colorectal Tumors and Related Metastatic Sites: Implications for Clinical Practice. Oncologist. 2008;13(12):1270–5.
46. Dergham ST, Dugan MC, Kucway R, Du W, Kamarauskiene DS, Vaitkevicius VK, et al. Prevalence and clinical significance of combined K-ras mutation and p53 aberration in pancreatic adenocarcinoma. Int J Pancreatol. 1997;21(2):127–43.
47. Galiana C, Lozano J ‐C, Bancel B, Nakazawa H, Yamasaki H. High frequency of ki‐ras amplification and p53 gene mutations in adenocarcinomas of the human esophagus. Mol Carcinog. 1995;14(4):286–93.
48. von Lintig FC, Dreilinger AD, Varki NM, Wallace AM, Casteel DE, Boss GR. Ras activation in human breast cancer. Breast Cancer Res Treat. 2000;.62(1):51–62.
49. Mascaux C, Iannino N, Martin B, Paesmans M, Berghmans T, Dusart M, et al. The role of RAS oncogene in survival of patients with lung cancer: A systematic review of the literature with meta-analysis. Br J Cancer. 2005;92(1):131–9.
50. Pretlow TP, Pretlow TG. Mutant KRAS in aberrant crypt foci (ACF): Initiation of colorectal cancer? Biochim Biophys Acta – Rev Cancer. 2005;1756(2):83–96.
51. Takayama T, Ohi M, Hayashi T, Miyanishi K, Nobuoka A, Nakajima T, et al. Analysis of K-ras, APC, and β-catenin in aberrant crypt foci in sporadic adenoma, cancer, and familial adenomatous polyposis. Gastroenterology. 2001;121(3):599–611.
52. Hecht JR, Douillard JY, Schwartzberg L, Grothey A, Kopetz S, Rong A, et al. Extended RAS analysis for anti-epidermal growth factor therapy in patients with metastatic colorectal cancer. Cancer Treat Rev. 2015;41(8):653–9.
53. Di Fiore F, Blanchard F, Charbonnier F, Le Pessot F, Lamy A, Galais MP, et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br J Cancer. 2007;96(8):1166–9.
54. Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-Type KRAS Is Required for Panitumumab Efficacy in Patients With Metastatic Colorectal Cancer. J Clin Oncol. 2008;26(10):1626–34.
55. Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras Mutations and Benefit from Cetuximab in Advanced Colorectal Cancer. N Engl J Med. 2008;359(17):1757–65.
56. Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer. 2009;101(4):715–21.
57. Irahara N, Baba Y, Nosho K, Shima K, Yan L, Dias-Santagata D, et al. NRAS Mutations Are Rare in Colorectal Cancer. Diagnostic Mol Pathol. 2010;19(3):157–63.
58. Sepulveda AR, Hamilton SR, Allegra CJ, Grody W, Cushman-Vokoun AM, Funkhouser WK, et al. Molecular Biomarkers for the Evaluation of Colorectal Cancer: Guideline From the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and the American Society of Clinical Oncology. J Clin Oncol. 2017;35(13):1453–86.
59. Allegra CJ, Rumble RB, Hamilton SR, Mangu PB, Roach N, Hantel A, et al. Extended RAS Gene Mutation Testing in Metastatic Colorectal Carcinoma to Predict Response to Anti–Epidermal Growth Factor Receptor Monoclonal Antibody Therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015. J Clin Oncol. 2016;34(2):179–85.
60. Markman B, Javier Ramos F, Capdevila J, Tabernero J. EGFR and KRAS in Colorectal Cancer. Adv Clin Chem. 2010;51(C):71–119.
61. Vijayaraghavan A, Efrusy MB, Göke B, Kirchner T, Santas CC, Goldberg RM. Cost-effectiveness of KRAS testing in metastatic colorectal cancer patients in the United States and Germany. Int J Cancer. 2012;131(2):438–45.
62. Behl AS, Goddard KAB, Flottemesch TJ, Veenstra D, Meenan RT, Lin JS, et al. Cost-Effectiveness Analysis of Screening for KRAS and BRAF Mutations in Metastatic Colorectal Cancer. JNCI J Natl Cancer Inst. 2012;104(23):1785–95.
63. Ozaki T, Nakagawara A. Role of p53 in cell death and human cancers. Cancers (Basel). 2011;3(1):994–1013.
64. Leslie A, Carey FA, Pratt NR, Steele RJC. The colorectal adenoma-carcinoma sequence. Br J Surg. 2002;89(7):845–60.
65. Béroud C, Soussi T. The UMD-p53 database: New mutations and analysis tools. Hum Mutat. 2003;21(3):176–81.
66. Sheng J, Sun H, Yu FB, Li B, Zhang Y, Zhu YT. The role of cyclooxygenase-2 in colorectal cancer. Int J Med Sci. 2020;17(8):1095–101.
67. Tanaka T, Watanabe T, Kazama Y, Tanaka J, Kanazawa T, Kazama S, et al. Chromosome 18q deletion and Smad4 protein inactivation correlate with liver metastasis: a study matched for T- and N- classification. Br J Cancer. 2006;95(11):1562–7.
68. Tanaka T, Watanabe T, Kazama Y, Tanaka J, Kanazawa T, Kazama S, et al. Loss of Smad4 protein expression and 18qLOH as molecular markers indicating lymph node metastasis in colorectal cancer-a study matched for tumor depth and pathology. J Surg Oncol. 2008;97(1):69–73.
69. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High Frequency of Mutations of the PIK3CA Gene in Human Cancers. Science. 2004;304(5670):554–554.
70. Domingo E, Church DN, Sieber O, Ramamoorthy R, Yanagisawa Y, Johnstone E, et al. Evaluation of PIK3CA mutation as a predictor of benefit from nonsteroidal anti-inflammatory drug therapy in colorectal cancer. J Clin Oncol. 2013;31(34):4297–305.
71. Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, et al. Aspirin Use, Tumor PIK3CA Mutation, and Colorectal-Cancer Survival. N Engl J Med. 2012;367(17):1596–606.
72. Kothari N, Kim R, Jorissen RN, Desai J, Tie J, Wong H-L, et al. Impact of regular aspirin use on overall and cancer-specific survival in patients with colorectal cancer harboring a PIK3CA mutation. Acta Oncol (Madr). 2015;54(4):487–92.
73. Waller A, Findeis S, Lee M. Familial Adenomatous Polyposis. J Pediatr Genet. 2016;05(02):078–83.
74. Dinarvand P, Davaro EP, Doan J V., Ising ME, Evans NR, Phillips NJ, et al. Familial adenomatous polyposis syndrome an update and review of extraintestinal manifestations. Arch Pathol Lab Med. 2019;143(11):1382–98.
75. Carr S, Casi A. Familial Adenomatous Polyposis. [Updated 2020 Nov 24]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; [Internet]. Treasure Island (FL): StatPearls Publishing; 2021.
76. Carneiro F, Lax SF, Arends MJ. Genetic tumour syndromes of the digestive system. In: Arends MJ, Fukayama M, Klimstra DS, Lam AKY, Nagtegaal ID, Odze RD, et al., editors. Digestive system tumors. 5th ed. Lyon: IARC Publications; 2019 Digestive system tumors. 5th ed.
77. Wu T-T, Kornacki S, Rashid A, Yardley JH, Hamilton SR. Dysplasia and Dysregulation of Proliferation in Foveolar and Surface Epithelia of Fundic Gland Polyps From Patients With Familial Adenomatous Polyposis. Am J Surg Pathol. 1998;22(3):293–8.
78. Arvanitis ML, Jagelman DG, Fazio VW, Lavery IC, McGannon E. Mortality in patients with familial adenomatous polyposis. Dis Colon Rectum. 1990;33(8):639–42.
79. Attard TM, Giglio P, Koppula S, Snyder C, Lynch HT. Brain tumors in individuals with Familial Adenomatous Polyposis: A cancer registry experience and pooled case report analysis. Cancer. 2007;109(4):761–6.
80. Findeisen P, Kloor M, Merx S, Sutter C, Woerner SM, Dostmann N, et al. T25 repeat in the 3’ untranslated region of the CASP2 gene: A sensitive and specific marker for microsatellite instability in colorectal cancer. Cancer Res. 2005;65(18):8072–8.
81. Baretti M, Le DT. DNA mismatch repair in cancer. Pharmacol Ther. 2018;189:45–62.
82. Yamamoto H, Imai K. Microsatellite instability: an update. Arch Toxicol. 2015.89(6):899–921.
83. Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710.
84. Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18(1):85–98.
85. Zhang Y, Yuan F, Presnell SR, Tian K, Gao Y, Tomkinson AE, et al. Reconstitution of 5’-directed human mismatch repair in a purified system. Cell. 2005;122(5):693–705.
86. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138(6):2073-2087.e3.
87. Li K, Luo H, Huang L, Luo H, Zhu X. Microsatellite instability: a review of what the oncologist should know. Cancer Cell Int. 2020;20(1):16.
88. Bhattacharya P, McHugh TW. Lynch Syndrome [Internet]. Treasure Island (FL): StatPearls Publishing; 2021.
89. Saridaki Z. Prognostic and predictive significance of MSI in stages II/III colon cancer. World J Gastroenterol. 2014;20(22):6809.
90. NCCN. NCCN Guidelines Version 2.2021 Colon Cancer. 2021
91. André T, Shiu K-K, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N Engl J Med. 2020;383(23):2207–18.
92. Lizardo DY, Kuang C, Hao S, Yu J, Huang Y, Zhang L. Immunotherapy efficacy on mismatch repair-deficient colorectal cancer: From bench to bedside. Biochim Biophys Acta – Rev Cancer. 2020;1874(2):188447.
93. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz H-J, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–91.
94. Yearsley M, Hampel H, Lehman A, Nakagawa H, de la Chapelle A, Frankel WL. Histologic features distinguish microsatellite-high from microsatellite-low and microsatellite-stable colorectal carcinomas, but do not differentiate germline mutations from methylation of the MLH1 promoter. Hum Pathol. 2006;37(7):831–8.
95. Greenson JK, Huang S-C, Herron C, Moreno V, Bonner JD, Tomsho LP, et al. Pathologic Predictors of Microsatellite Instability in Colorectal Cancer. Am J Surg Pathol. 2009;33(1):126–33.
96. Kakar S, Shi C, Berho ME, Driman DK, Fitzgibbons P, Frankel WL, et al. Protocol for the Examination of Specimens From Patients With Primary Carcinoma of the Colon and Rectum, Colon Rectum 4.0.1.0. 2017.
97. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248–57.
98. Vilar E, Gruber SB. Microsatellite instability in colorectal cancer—the stable evidence. Nat Rev Clin Oncol. 2010.7(3):153–62.
99. Yuan L, Chi Y, Chen W, Chen X, Wei P, Sheng W, et al. Immunohistochemistry and microsatellite instability analysis in molecular subtyping of colorectal carcinoma based on mismatch repair competency. Int J Clin Exp Med. 2015;8(11):20988–1000.
100. Fleming M, Ravula S, Tatishchev SF, Wang HL. Colorectal carcinoma: Pathologic aspects. J Gastrointest Oncol. 2012;3(3):153–73.
101. Chen M-L, Chen J-Y, Hu J, Chen Q, Yu L-X, Liu B-R, et al. Comparison of microsatellite status detection methods in colorectal carcinoma. Int J Clin Exp Pathol. 2018;11(3):1431–8.
102. Chen W, Frankel WL. A practical guide to biomarkers for the evaluation of colorectal cancer. Mod Pathol. 2019;32(S1):1–15.
103. Rosty C, Clendenning M, Walsh MD, Eriksen S V, Southey MC, Winship IM, et al. Germline mutations in PMS2 and MLH1 in individuals with solitary loss of PMS2 expression in colorectal carcinomas from the Colon Cancer Family Registry Cohort. BMJ Open. 2016;6(2):e010293.
104. Saeed OAM, Mann SA, Luchini C, Huang K, Zhang S, Sen JD, et al. Evaluating mismatch repair deficiency for solid tumor immunotherapy eligibility: immunohistochemistry versus microsatellite molecular testing. Hum Pathol. 2021;115:10–8.
105. Dedeurwaerdere F, Claes KB, Van Dorpe J, Rottiers I, Van der Meulen J, Breyne J, et al. Comparison of microsatellite instability detection by immunohistochemistry and molecular techniques in colorectal and endometrial cancer. Sci Rep. 2021;11(1):12880.
106. Hempelmann JA, Scroggins SM, Pritchard CC, Salipante SJ. MSIplus for Integrated Colorectal Cancer Molecular Testing by Next-Generation Sequencing. J Mol Diagnostics. 2015;17(6):705–14.
107. Xiao J, Li W, Huang Y, Huang M, Li S, Zhai X, et al. A next-generation sequencing-based strategy combining microsatellite instability and tumor mutation burden for comprehensive molecular diagnosis of advanced colorectal cancer. BMC Cancer. 2021.21(1):282.
108. Lee Y, Lee JA, Park HE, Han H, Kim Y, Bae JM, et al. Targeted next-generation sequencing-based detection of microsatellite instability in colorectal carcinomas. Galli A, editor. PLoS One. 2021;16(2):e0246356.
109. Ulahannan D, Kovac MB, Mulholland PJ, Cazier J-B, Tomlinson I. Technical and implementation issues in using next-generation sequencing of cancers in clinical practice. Br J Cancer. 2013;109(4):827–35.
110. Del Vecchio F, Mastroiaco V, Di Marco A, Compagnoni C, Capece D, Zazzeroni F, et al. Next-generation sequencing: recent applications to the analysis of colorectal cancer. J Transl Med. 2017;15(1):246.
111. Murphy KM, Zhang S, Geiger T, Hafez MJ, Bacher J, Berg KD, et al. Comparison of the microsatellite instability analysis system and the Bethesda panel for the determination of microsatellite instability in colorectal cancers. J Mol Diagnostics. 2006;8(3):305–11.
112. Steinke V, Engel C, Büttner R, Schackert HK, Schmiegel WH, Propping P. Hereditary Nonpolyposis Colorectal Cancer (HNPCC)/Lynch Syndrome. Dtsch Arztebl Int. 2013;110(3):32-8.
113. Abedalthagafi M. Constitutional mismatch repair-deficiency: current problems and emerging therapeutic strategies. Oncotarget. 2018;9(83):35458–69.
114. Ligtenberg MJL, Kuiper RP, Geurts van Kessel A, Hoogerbrugge N. EPCAM deletion carriers constitute a unique subgroup of Lynch syndrome patients. Fam Cancer. 2013;12(2):169–74.
115. Jin B, Li Y, Robertson KD. DNA Methylation: Superior or Subordinate in the Epigenetic Hierarchy? Genes Cancer. 2011;2(6):607–17.
116. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36.
117. Leggett B, Whitehall V. Role of the Serrated Pathway in Colorectal Cancer Pathogenesis. Gastroenterology. 2010;138(6):2088–100.
118. Mojarad EN, Kuppen PJK, Aghdaei HA, Zali MR. The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol Hepatol from Bed to Bench. 2013;6(3):120–8.
119. Gonzalez RS, Washington K, Shi C. Current applications of molecular pathology in colorectal carcinoma. Appl Cancer Res. 2017;37(1):1–13.
120. Thibodeau SN, French AJ, Cunningham JM, Tester D, Burgart LJ, Roche PC, et al. Microsatellite instability in colorectal cancer: different mutator phenotypes and the principal involvement of hMLH1. Cancer Res. 1998;58(8):1713–8.
121. De Palma FDE, D’argenio V, Pol J, Kroemer G, Maiuri MC, Salvatore F. The molecular hallmarks of the serrated pathway in colorectal cancer. Cancers (Basel). 2019;11(7):3–5.
122. Ogino S, Goel A. Molecular Classification and Correlates in Colorectal Cancer. J Mol Diagnostics. 2008;10(1):13–27.
123. Caputo, Santini, Bardasi, Cerma, Casadei-Gardini, Spallanzani, et al. BRAF-Mutated Colorectal Cancer: Clinical and Molecular Insights. Int J Mol Sci. 2019;20(21):5369.
124. Ducreux M, Chamseddine A, Laurent-Puig P, Smolenschi C, Hollebecque A, Dartigues P, et al. Molecular targeted therapy of BRAF -mutant colorectal cancer. Ther Adv Med Oncol. 2019;11:1758835919856494.
125. Zhu H, Blake S, Kusuma FK, Pearson RB, Kang J, Chan KT. Oncogene-induced senescence: From biology to therapy. Mech Ageing Dev. 2020;187:111229.
126. Kim JH, Kang GH. Evolving pathologic concepts of serrated lesions of the colorectum. J Pathol Transl Med. 2020;54(4):276–89.
127. Kim SY, Kim T Il. Serrated neoplasia pathway as an alternative route of colorectal cancer carcinogenesis. Intest Res. 2018;16(3):358.
128. Luu L-J, J. Price T. BRAF Mutation and Its Importance in Colorectal Cancer. In: Advances in the Molecular Understanding of Colorectal Cancer. IntechOpen; 2019 Advances in the Molecular Understanding of Colorectal Cancer.
129. Venderbosch S, Nagtegaal ID, Maughan TS, Smith CG, Cheadle JP, Fisher D, et al. Mismatch Repair Status and BRAF Mutation Status in Metastatic Colorectal Cancer Patients: A Pooled Analysis of the CAIRO, CAIRO2, COIN, and FOCUS Studies. Clin Cancer Res. 2014;20(20):5322–30.
130. Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer. N Engl J Med. 2019;381(17):1632–43.
131. Oh K, Redston M, Odze RD. Support for hMLH1 and MGMT silencing as a mechanism of tumorigenesis in the hyperplastic–adenoma-carcinoma (serrated) carcinogenic pathway in the colon. Hum Pathol. 2005;36(1):101–11.
132. Phipps AI, Limburg PJ, Baron JA, Burnett-Hartman AN, Weisenberger DJ, Laird PW, et al. Association Between Molecular Subtypes of Colorectal Cancer and Patient Survival. Gastroenterology. 2015;148(1):77-87.e2.
133. Phipps AI, Alwers E, Harrison T, Banbury B, Brenner H, Campbell PT, et al. Association Between Molecular Subtypes of Colorectal Tumors and Patient Survival, Based on Pooled Analysis of 7 International Studies. Gastroenterology. 2020;158(8):2158-2168.e4.